Seguindo a linha de convidados especiais, o blog da A.Flux dessa semana conta com a contribuição da Dra. Nelbe Nesi. O tema tratado por ela é bastante relevante e esclarece muitas dúvidas sobre a Fibrose Cística.
Boa Leitura!
—————
Você conhece a Fibrose Cística?
Também conhecida como mucoviscidose, a Fibrose Cística é uma doença rara, genética, autossômica, recessiva, crônica e, muitas vezes, letal que acomete vários órgãos. Uma de suas características é a perda acentuada de eletrólitos pelo suor, que pode levar a desidratação e gera um de seus sinais mais importantes: o suor salgado. Logo, ficou conhecida como a doença do beijo salgado. Embora as manifestações respiratórias sejam responsáveis por 90% da morbidade e mortalidade na Fibrose Cística, os outros acometimentos da doença levam a importantes limitações físicas, podendo gerar impacto na qualidade de vida e na capacidade funcional do indivíduo.
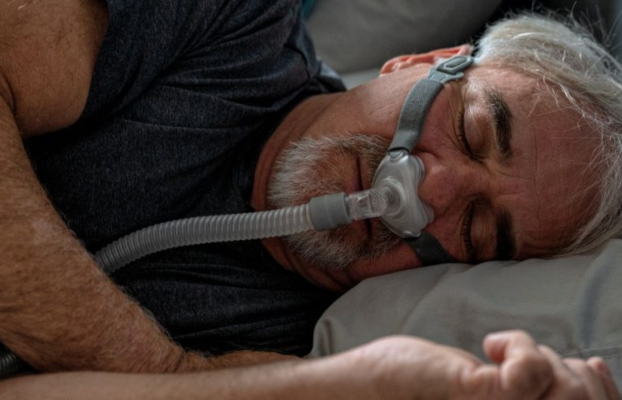
Na Fibrose Cística, as secreções corporais se apresentam mais viscosas, impactando muitos órgãos. No sistema respiratório, o muco viscoso fica acumulado nas vias aéreas e nos pulmões, gerando inflamação e facilitando a colonização de bactérias que agravam ainda mais o quadro. O acúmulo de muco espesso no sistema respiratório gera tosse crônica e, num estágio mais avançado, destruição pulmonar. Portanto, na fase mais tardia da doença, é comum o uso contínuo de tecnologias como a ventilação não-invasiva e a suplementação de oxigênio.
O tratamento da doença pulmonar crônica presente na Fibrose Cística se dá através de nebulizações específicas e sessões de fisioterapia respiratória e exercício físico inseridos na rotina diária dos pacientes. Também é comum o uso de antibióticos orais na tentativa de erradicar as bactérias colonizadas na secreção das vias aéreas e reduzir a inflamação.
Contudo, não é só o sistema respiratório que se encontra acometido. O sistema digestório também se encontra prejudicado pelo acúmulo de muco viscoso no pâncreas e a alteração de secreção de bicarbonato nos intestinos. O acúmulo de muco nos ductos pancreáticos gera obstrução dos mesmos, prejudicando a secreção das enzimas pancreáticas que auxiliam na digestão. A redução ou até mesmo ausência das enzimas pancreáticas, associada à alteração no pH intestinal devido ao distúrbio na secreção de bicarbonato, levam à má absorção intestinal que, na maioria dos casos, tornam as fezes muito oleosas e geram desnutrição crônica nestes pacientes. O tratamento das alterações do sistema digestório é baseado na suplementação de enzimas pancreáticas e vitaminas importantes para o organismo. Além disso, é comum a prescrição de dietas hipercalóricas pelo nutricionista, na tentativa de minimizar a desnutrição no indivíduo com Fibrose Cística.
O diagnóstico da Fibrose Cística é feito através do teste do pezinho e confirmado pelo teste do suor realizado no centro de referência para a doença. Também é possível diagnosticar a Fibrose Cística pelo exame genético, onde é necessário haver duas mutações patológicas no gene CFTR (Cystic Fibrosis Transmenbrane Conductance Regulator).
Ao longo das décadas, inúmeros estudos vêm sendo realizados, proporcionando o surgimento de novos tratamentos para a Fibrose Cística. Portanto, o diagnóstico precoce é de extrema importância dada a característica crônica e progressiva da doença. Além disso, com o desenvolvimento da terapia gênica, conhecer a mutação genética causadora da patologia é imprescindível para a prescrição dessa nova tecnologia.
Assim, se você, seu filho (a) ou alguém que você conhece apresenta suor salgado, fezes oleosas associadas ou não à diarreia e/ou tosse crônica, procure um pediatra ou clínico. O diagnóstico precoce pode fazer total diferença no prognóstico da doença.
——
REFERÊNCIAS:
Instituto Brasileiro de Atenção à Fibrose Cística
Associação Carioca de Amigos da Mucoviscidose – Rio de Janeiro.
Grupo Brasileiro de Estudos de Fibrose Cística